The EU AI Act classifies most health applications as high-risk — what that means for diagnostics, treatment, and research
Artificial intelligence entered European healthcare policy language long before it entered clinical practice at any significant scale. The European Commission’s first coordinated AI plan, published in 2018, identified health as a priority application domain. Horizon Europe funding has supported dozens of AI-in-health research projects. Regulatory frameworks have been drafted, consulted upon, and adopted. And yet the gap between political ambition and clinical deployment — between the AI systems that appear in funding proposals and those that are actually used by clinicians in daily practice — remains substantial. Understanding that gap, and the regulatory architecture that now governs the space, requires distinguishing between what AI systems can demonstrably do in healthcare and what they are permitted — and resourced — to do in European health systems.
The EU AI Act, which entered into force in August 2024, represents the most consequential regulatory development in this space. It classifies AI systems used in medical contexts — diagnostic software, clinical decision support tools, AI-enabled medical devices — largely as high-risk, triggering a compliance framework that includes mandatory conformity assessment, data quality requirements, human oversight obligations, and transparency requirements that did not previously apply to software medical devices under the Medical Devices Regulation alone. The implications are significant for an ecosystem that was, until August 2024, operating in a regulatory space that the MDR addressed incompletely and that general data protection law did not fully reach.
The AI Act’s High-Risk Classification: What It Means for Health AI
The AI Act’s Annex III lists the categories of AI system classified as high-risk. AI systems intended to be used as safety components of medical devices, or which are themselves medical devices subject to conformity assessment under the Medical Devices Regulation (MDR) or the In Vitro Diagnostic Regulation (IVDR), fall within the high-risk category. In practice, this covers a wide range of health AI applications: diagnostic imaging analysis tools that identify tumours or haemorrhages; clinical decision support systems that recommend treatment protocols; AI systems for patient triage; and software that interprets clinical test results.
The compliance obligations attached to high-risk classification are substantive. AI systems must be trained on high-quality datasets that are representative of the populations they will be deployed in — a requirement that directly implicates the data governance infrastructure that the EHDS data infrastructure under development will eventually provide, but does not yet consistently offer. They must be designed with human oversight mechanisms that allow clinical users to understand and override AI outputs. They must maintain technical documentation sufficient to support conformity assessment and post-market surveillance. And they must meet transparency requirements ensuring that affected individuals — patients — know when AI systems have been involved in decisions affecting them.
The implementation timeline gives high-risk AI systems 36 months from the Act’s entry into force before compliance obligations fully apply — meaning the main obligations will apply from August 2027 for AI systems that are also regulated medical devices under MDR or IVDR. The Commission was required to publish guidelines on the practical implementation of high-risk classifications, including a list of use cases falling within and outside the high-risk category, by February 2026. That guidance is consequential: the boundary between high-risk and non-high-risk classification determines whether an AI system faces the full compliance framework or a lighter-touch obligations set.
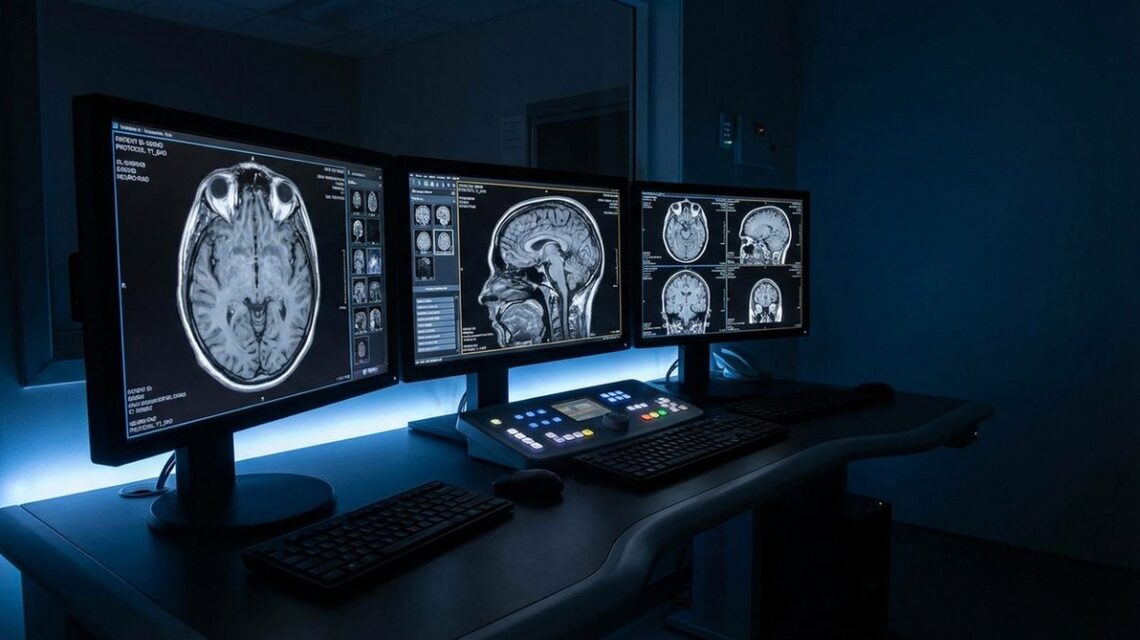
Diagnostics: Where AI Deployment Is Most Advanced
Diagnostic imaging analysis is the domain in which AI deployment in clinical medicine is most advanced, and where the evidence base is strongest. AI systems trained to identify radiological findings — pulmonary nodules in CT scans, diabetic retinopathy in fundus photographs, skin lesions in dermatological images — have demonstrated performance comparable to specialist clinicians in controlled study conditions across multiple peer-reviewed publications. Several CE-marked AI diagnostics tools are in commercial deployment across EU member states.
The clinical reality is more qualified than the published benchmarks suggest. Studies reporting AI diagnostic performance comparable to radiologists or dermatologists typically evaluate performance on curated datasets — collections of cases where the ground truth is established — under conditions that may not reflect the variability and image quality of clinical practice. When AI diagnostic tools are evaluated in real-world deployment, performance frequently declines relative to controlled study conditions. The reasons are multiple: the demographic and disease characteristics of a tool’s training population may not match the characteristics of the clinical population in which it is deployed, creating bias that manifests as systematic under- or over-diagnosis for specific groups.
The European Medicines Agency reached a significant milestone in March 2025 when its human medicines committee issued a qualification opinion on the AIM-NASH tool — AI software designed to support pathologists in analysing liver biopsy scans for the severity of metabolic dysfunction-associated steatohepatitis (MASH), a condition relevant to clinical trial design for hepatology treatments. The qualification opinion represents regulatory acknowledgment that AI-based analysis can be used as an innovative development methodology in the medicines approval process — a signal of increasing institutional confidence in specific, well-evaluated AI applications, as distinct from the broader class of health AI tools.
Drug Discovery and Research: AI’s Faster Track to Impact
If clinical AI deployment faces the friction of regulated health systems and conservative clinical adoption, AI in pharmaceutical research faces different but arguably more tractable challenges. Drug discovery is a domain where AI has found commercially productive applications more rapidly than in clinical medicine, in part because the feedback loops are more legible — computational predictions of molecular properties or biological activity can be tested in laboratory settings on timescales that allow iterative learning — and in part because the regulatory pathway, while complex, is less directly contingent on real-world clinical data from human populations.
European examples illustrate the range of AI application in drug discovery. Budapest-based Turbine uses mechanistic cell models to screen genetic perturbations in silico at scale, surfacing potential synthetic-lethal targets for oncology research. French company Iktos applies generative algorithms to design synthesisable small molecules, compressing timelines for molecular design from months to hours. Dublin’s Nuritas uses AI to discover bioactive peptides. Denmark’s Evaxion Biotech deploys an AI-Immunology platform to propose personalised vaccine antigens and cancer immunotherapies. These companies represent a European AI drug discovery ecosystem that, while smaller than the US cluster centred on companies like Recursion or Insilico Medicine, is scientifically productive and growing.
The European Medicines Agency has been developing its framework for evaluating AI-generated evidence in regulatory submissions, recognising that medicines developed with significant AI involvement in target identification, molecular design, or clinical trial design will increasingly reach the approval process. A 2024 EMA horizon-scanning report on AI and machine learning in the medicines lifecycle identified the key regulatory questions the agency anticipates managing: how to evaluate the validity of AI-generated biological hypotheses, how to assess the representativeness of AI training datasets, and how to integrate AI-assisted analysis into established standards of evidence.
The Bias and Explainability Challenge
Two technical challenges cut across all health AI application domains and are central to the EU regulatory framework’s design: bias and explainability. Both are recognised in the AI Act as issues that high-risk AI systems must address; both are genuine limitations on the current state of health AI that the regulatory framework can identify more clearly than it can resolve.
Bias in health AI arises from multiple sources, of which training data composition is the most frequently discussed but not the only relevant factor. If an AI diagnostic system is trained predominantly on data from European or North American clinical populations — the source of most large medical imaging datasets — its performance for patients from populations underrepresented in the training data may be systematically lower. This bias pattern has been documented in dermatological AI, where published studies have found that tools trained predominantly on images of lighter-skinned patients perform less reliably for darker-skinned patients. It has been identified in cardiac diagnostic AI, in predictive models for hospital readmission, and in clinical risk scoring tools. The AI Act’s requirement for high-quality, representative training data is a response to this documented pattern; ensuring that the requirement is met for health AI systems deployed across EU populations with significant demographic diversity is an implementation challenge that the EHDS data infrastructure is designed to support, but will not be able to address fully before the Act’s compliance obligations apply.
Explainability — the capacity to provide human-understandable accounts of how an AI system reached a specific output — is both a regulatory requirement and a clinical necessity. A clinician using an AI diagnostic tool needs to understand whether the system’s recommendation is consistent with the imaging features they observe, and where the AI’s assessment diverges from their own clinical judgment. Deep learning models, the dominant architecture in medical imaging AI, are intrinsically opaque: the features driving their predictions are distributed across millions of parameters in ways that do not translate straightforwardly into clinical reasoning. Explainable AI techniques — saliency maps that highlight image regions influencing a model’s prediction, counterfactual explanations that describe how an output would change with different inputs — have been developed and are improving, but they provide approximations of explainability rather than the mechanistic transparency that the term might imply.
EU vs. US: Divergent Regulatory Philosophies
The contrast between the EU’s regulatory approach to health AI and the approach taken in the United States is instructive for understanding what the EU framework will and will not achieve. The US approach, managed primarily through the FDA’s Digital Health Center of Excellence, has been to develop guidance through iterative engagement with industry, with a generally permissive posture toward novel AI applications, a preference for post-market surveillance over pre-market requirements, and a faster approval pathway for AI-enabled medical devices that has resulted in several hundred AI-enabled medical device authorisations.
The EU’s approach — high-risk classification, mandatory conformity assessment, strict data quality requirements, human oversight obligations — is more prescriptive and, in the near term, more compliance-intensive for developers. It also reflects a different political philosophy about where the burden of proof should lie: in the EU framework, a health AI system must demonstrate that it meets the required standards before deployment; in the US framework, the mechanism for catching problems is more heavily weighted toward post-market detection. The tradeoff involves innovation pace against risk management — a tension that neither framework resolves entirely, and that different member states within the EU framework are navigating with varying degrees of enthusiasm for enforcement.
For workforce readiness for digital health, the EU’s regulatory approach creates particular demands. Clinicians using high-risk AI systems must have sufficient understanding of the system’s capabilities and limitations to exercise meaningful oversight — a requirement that implies training needs going well beyond current digital health curricula in most EU medical education systems. The AI Act’s human oversight requirements cannot be met by deploying AI systems to clinical users who lack the competence to critically evaluate AI outputs, which means the regulatory framework’s implementation depends on workforce development that is, in most member states, still in early stages.
The intersection of health AI with the EU’s broader data and digital governance agenda — the EHDS, the AI Act, the Data Governance Act, and the Data Act — creates a regulatory architecture that is comprehensive in scope and complex in practice. Companies developing health AI for the EU market face compliance obligations across multiple regulatory instruments; health systems deploying AI must navigate procurement, conformity assessment, clinical integration, and post-market surveillance requirements simultaneously. Whether that complexity serves the goal of safe and effective health AI deployment, or primarily raises barriers that reduce the available tool set for European health systems, is a question the next several years of implementation will begin to answer.



